Leitfaden Genetik
Zellen sind die grundlegenden Arbeitseinheiten jedes lebenden Systems. Alle Anweisungen zur Regelung ihrer Aktivitäten sind in der DNA enthalten, die sich in jedem einzelnen Zellkern befindet.
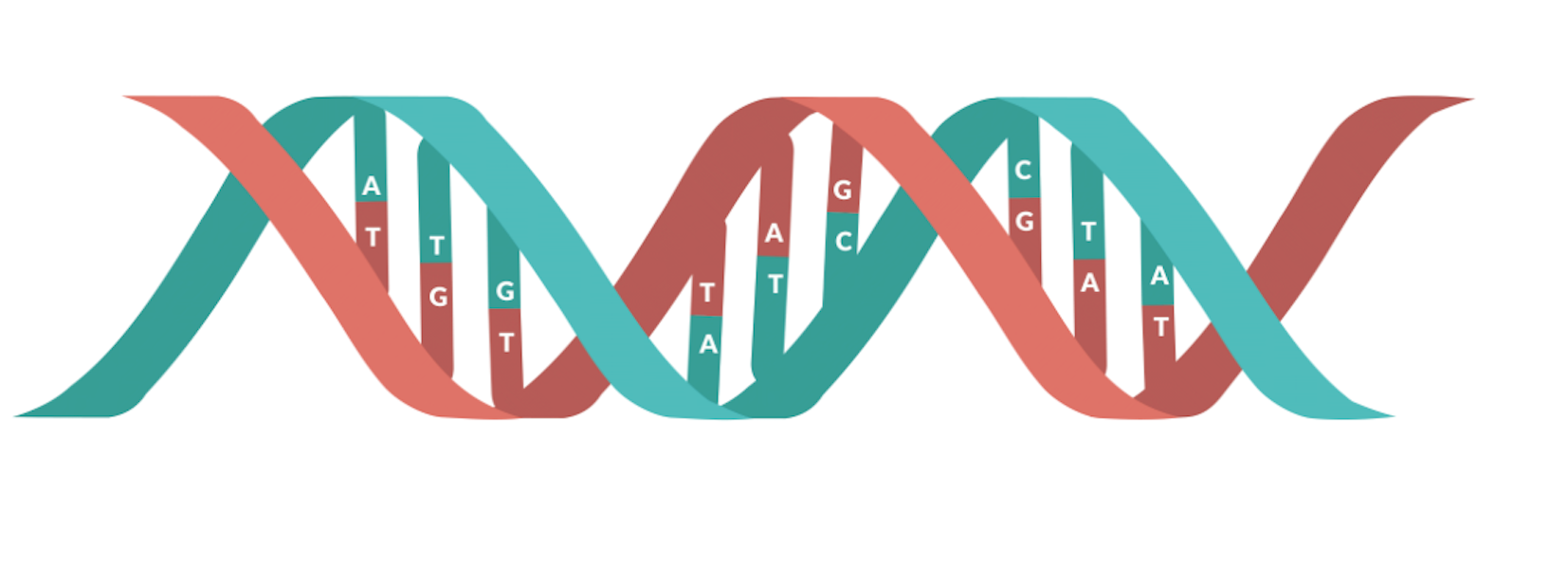
Die DNA aller Organismen, von den kleinsten Bakterien bis hin zum Menschen, besteht aus den gleichen chemischen und physikalischen Bausteinen. Die DNA ist wie eine in sich gedrehte Leiter aufgebaut und hat Sprossen zwischen den Seitenteilen, die wie ein Reißverschluss „aufgezogen“ werden können. Die Sprossen setzen sich aus Nukleotidbasen zusammen, die A(Adedin),T(Thymian),C(Cytosin), und G(Guanin) genannt werden. A bildet immer ein Paar mit T, und C bindet immer an G. Diese Ordnung bildet die exakten Anweisungen zum Aufbau eines speziellen Organismus mit seinen eigenen, unverwechselbaren Eigenschaften.
Das Genom ist das vollständige DNA-Set eines Organismus. Genome variieren beachtlich in ihrer Größe: Das kleinste bekannte Genom eines frei lebenden Organismus (ein Bakterium) enthält etwa 600.000 DNA-Basenpaare, wogegen menschliche und Mausgenome um die drei Milliarden haben. Jede menschliche Zelle enthält das komplette Genom.
Die DNA des menschlichen Genoms ist in 22 Chromosomen und 1 Geschlechtsbestimmendes Chromosomen Paar untergebracht, und jedes davon enthält viele Gene. Die X- und Y-Chromosomen bestimmen das Geschlecht des Säuglings. Mädchen haben zwei X-Chromosomen und kein Y, Jungen haben ein X und ein Y. Das MECP2-Gen befindet sich auf dem X-Chromosom. Mädchen mit Rett haben entsprechend zwei MECP2-Gene, ein mutiertes und eins, das sich normal verhält.
Obwohl den Genen eine Menge Aufmerksamkeit gilt, sind es doch die Proteine, die durch ihren Code gebildet werden und die die meisten Lebensfunktionen steuern sowie den Großteil der Zellstrukturen aufbauen.
Proteine sind große und komplexe Moleküle, die aus zahlreichen Untereinheiten, den so genannten Aminosäuren bestehen. Chemische Merkmale, in denen sich die 20 verschiedenen Aminosäuren unterscheiden, lassen die Proteinketten sich in spezifischen dreidimensionalen Strukturen anordnen, was wiederum ihre besonderen Funktionen in der Zelle definiert.

Was bedeutet die Mutation bei meinem Kind?
Eine Genmutation ist eine dauerhafte Veränderung der DNA-Sequenz. Mutationen variieren bezüglich ihrer Größe von nur einer Nukleotidbase bis hin zu einem großen Segment der DNA, das viele Gene enthält.
Eine Mutation kann verursachen, dass das Protein schlecht funktioniert oder völlig fehlt, indem sie die Information des Gens für den Aufbau eines Proteins verändert. Bei Rett führt eine Mutation des MECP2-Gens dazu, dass das MeCP2-Protein ebenfalls mutiert.
Es ist wichtig festzuhalten, dass Gene selbst keine Krankheit verursachen – genetische Störungen rühren von Mutationen her, die ein Gen unangemessen funktionieren lassen. Beispielsweise meinen die Leute eine mutierte Version des MECP2-Gens, wenn sie vom „Rett-Syndrom-Gen“ sprechen. Dieses löst die Störung aus. Alle Menschen, auch die ohne RETT, haben ein MECP2-Gen.
Die Befunde Ihres Kindes enthalten normalerweise zwei Informationen – eine Zahl, die sich auf die mutierte Nukleotidbase bezieht, und eine Zahl, die der damit verbundenen Veränderung in der Aminosäurensequenz entspricht.
Man könnte Ihnen auch sagen, dass die Mutation heterozygot ist, was bedeutet, dass Ihre Tochter ein normales X-Chromosom hat und ein X-Chromosom mit einem mutierten MECP2. Jungen mit Mutationen am MECP2 sind homozygot, weil sie nur ein X-Chromosom haben. Unten finden Sie ein paar Beispiele für typische Mutationen.
Missense-Mutation (Die Proteine codieren für eine falsche Aminosäure)
Dieser Typ Mutation ist die Veränderung in einem DNA-Basenpaar, was dazu führt, dass eine Aminosäure durch eine andere ersetzt wird.
Beispiel: 473 C → T / T158M
Die erste Zahl bezieht sich auf die mutierte Nukleotidbase. In diesem besonderen Fall sollte Nukleotidbase Nummer 473 eine Cytosin-Nukleotidbase sein, doch wegen der Mutation befindet sich dort nun Thymin.
Dies führt dazu, dass eine andere Aminosäure kodiert wird. Bei Aminosäure Nummer 158 (man teilt 473 durch 3, um zur Nummer der Aminosäure zu gelangen, weil immer 3 Basen eine Aminosäure kodieren) sollte Threonin sein, doch jetzt findet man dort Methionin. Diese einfache Veränderung nur einer Aminosäure von fast 500 macht das Protein dysfunktional.
Nonsense-Mutation
Eine Nonsense-Mutation ist ebenfalls eine Veränderung an einem DNA-Basenpaar. Doch anstatt eine Aminosäure durch eine andere zu ersetzen, signalisiert die DNA-Sequenz der Zelle frühzeitig, den Proteinaufbau einzustellen. Diese Art der Mutation führt zu einem verkürzten Protein, das möglicherweise unzureichend oder gar nicht funktioniert.
Beispiel: R168X (Das X bezeichnet eine Mutation, die zu einem frühzeitig gekürzten Protein führt.)
Insertion
Eine Insertion verändert die Anzahl der DNA-Basen in einem Gen, indem ein Stück DNA hinzugefügt wird, was ein dysfunktionales Protein hervorbringt.
Beispiel: 620insT (An der Nukleotidbase Nummer 620 wurde ein zusätzliches Thymin eingefügt.)
Löschung
Eine Löschung ändert die Anzahl der DNA-Basen im Gen, indem ein Stück DNA entfernt wird. Die gelöschte DNA verändert die Funktion des daraus hervorgehenden Proteins.
Beispiel: 803del4 (An Nukleotidbase Nummer 803 wurden vier Basen gelöscht.)
Leserastermutation
Diese Art Mutation tritt auf, wenn die Hinzufügung oder der Verlust von DNA-Basen das Leseraster eines Gens ändert. Ein Leseraster besteht aus Gruppen von 3 Basen, von denen je eine eine Aminosäure kodiert. Eine Leserastermutation verschiebt die Anordnung dieser Basen und ändert so den Code für Aminosäuren. Das so hervorgebrachte Protein funktioniert normalerweise nicht. Insertionen, Löschungen und Dopplungen können Leserastermutationen sein. Die Beispiele für 620insT und 803del4 oben basieren beide auf Leserastermutationen.
Kann ich die Symptome meines Kindes anhand der Mutation vorhersagen?
Eine wichtige Variable für die Symptomstärke ist die X-Deaktivierung. Frauen haben zwei X-Chromosomen, Männer nur eins. Damit Männer und Frauen die gleiche Menge an genetischem Material haben, müssen Frauen eins ihrer X-Chromosomen in jeder Zelle stumm schalten. Die meisten Frauen haben ein zufälliges Deaktivierungsmuster, das etwa 50% eines X und 50% des anderen aktiviert. Es kann jedoch aus bisher unbekannten Gründen dazu kommen, dass in einem weiblichen Körper ein X das andere überlagert.
Wenn ein Mädchen mit RETT bevorzugt das X-Chromosom mit dem mutierten MECP2 deaktiviert, kommt es zu weniger ernsten Symptomen. Passiert das Gegenteil, hat das Kind möglicherweise schwerwiegendere Symptome. Obwohl das X-Deaktivierungsmuster Ihrer Tochter im Blut getestet werden kann, sagt dies noch nichts über das entsprechende Muster in ihrem Gehirn, und entsprechend ist der Nutzen des Tests an diesem Punkt begrenzt.
Eine andere Variable ist die persönliche genetische Zusammensetzung des Kindes. Es gibt Personen mit Mutationen an MECP2 und normaler X-Chromosom-Überlagerung, die keine Rett-Symptome haben. Wahrscheinlich haben diese Personen Mutationen an anderen Genen, die einen Schutz gegen ihr mutiertes MECP2 bieten. Der RSRT finanziert gegenwärtig ein Projekt an solchen Modifikatorgenen.
Ein paar Studien, die eine Verbindung zwischen spezifischen Mutationen und Symptomen herstellen wollten, zeigen aktuell variierende Ergebnisse. Einige sehr allgemeine Schlussfolgerungen besagen:
- Die R133C-Mutation ist üblicherweise nicht so schwerwiegend
- Verkürzungsmutationen scheinen mit Atemstörungen einherzugehen, während Skoliose eher bei Personen mit Missense-Mutationen auftrat.
- Missense-Mutationen scheinen mildere Symptome zu verursachen
- Frühe Verkürzungsmutationen führen zu ernsteren Auswirkungen als späte Verkürzungs- oder Missense-Mutationen
Bitte beachten Sie, dass diese Verallgemeinerungen auf großen exemplarischen Mustern basieren. Schlussfolgerungen im Bezug auf die Mutation Ihres Kindes auf einer Fall-zu-Fall-Basis eignen sich nicht für zuverlässige Vorhersagen. Darüber hinaus wissen wir nicht, welche anderen Faktoren und Umwelteinflüsse die Wirkung einer spezifischen Mutation beeinflussen.

Woher kommt die Mutation?
Die große Mehrheit der Mutationen ist sporadisch, nicht familiär und entstammt einem mutierten Spermium. In solchen Fällen fällt der Test an einem Vater auf MECP2-Mutationen negativ aus, weil keine seiner Zellen Mutationen enthält, außer dem einzelnen Spermium, das die Eizelle befruchtet hat, aus der seine Tochter hervorging.
Bei Familien, in denen MECP2-Mutationen bei mehr als einem Familienmitglied nachgewiesen wurden, gibt es zwei mögliche Erklärungen.
- Die Mutter hat eine MECP2-Mutation, aber ihr X-Deaktivierungsmuster überlagert und so hat sie keine Symptome.
- Die Mutter hat Mutationen in ihren Eizellen, aber nicht in anderem Gewebe. Daher ist sie komplett symptomfrei, es besteht aber eine 50prozentige Chance, dass sie die Mutation an ihre Nachkommen vererbt. Dies bezeichnet man als Keimbahnmosaizismus.
Können Männer das Rett-Syndrom bekommen?
Auch wenn das selten vorkommt, können Männer Mutationen am MECP2 haben. Es gibt drei Szenarien, die zu einer männlichen Person mit RETT führen können.
- Ein Junge hat das Klinefelter-Syndrom (was einmal bei 1.000 männlichen Geburten vorkommt) und wird mit einem zusätzlichen X-Chromosom geboren (XXY). Eins der X-Chromosome hat die Mutation, das andere nicht. Diese Jungen haben Symptome, die denen bei Mädchen mit RETT ähnlich sind.
- Eine Mutation kommt nicht vom Spermium oder der Eizelle, sondern passiert auf einer etwas späteren Stufe, wie etwa der 20-Zellenstufe (Blastula). Wenn eine der Zellen mutiert, werden auch die daraus hervorgehenden Zellen mutieren, während die verbleibenden 19 Zellen und deren Folgezellen dies nicht tun. Dies nennt man somatischen Mosaizismus, und der Phänotyp ähnelt dem eines Mädchens mit RETT, weil ein somatischer Mosaizismus ähnlich funktioniert wie die X-Deaktivierungsmuster bei Mädchen.
- Ein Junge wird mit dem typischen XY-Karotyp geboren und hat Mutationen am MECP2 in allen seinen X-Chromosomen. Da der Junge keine intakte Kopie hat, um das mutierte Gen abzumildern, sind seine Symptome schwerwiegend und führen sehr wahrscheinlich zu einem frühen Tod. Es gibt aber aus bisher unbekannten Gründen auch Jungen mit „milderen“ Mutationen.
Interessanterweise beschrieb ein Artikel kürzlich eine Gruppe männlicher Personen mit zwei Kopien des MECP2 auf ihrem X-Chromosom, bei denen entsprechend nicht zu viel MeCP2-Protein produziert wird. Die Symptome der Jungen waren: Hypotonie, Entwicklungsverzögerung, keine Sprech- und Gehfähigkeit, Krämpfe, wiederkehrende Infektionen und spastische Lähmungen beim Aufwachsen. Bei diesen Fällen kamen die Mutationen am MECP2-Gen von Müttern mit stark überlagernder X-Deaktivierung, die aus unbekannter Ursache keine Symptome hatten.
Sollten Familienmitglieder getestet werden?
Eltern, Geschwister und andere Verwandte testen zu lassen ist eine persönliche Entscheidung, die jede Familie treffen muss. Die Entscheidung, ob Eltern sich testen lassen, ist stark beeinflusst von Entscheidungen zur Familienplanung.
Eltern einer Tochter mit einer MECP2-Mutation
Die große Mehrheit (99,5%) der RETT-Fälle treten einzeln innerhalb einer Familie auf und sind auf eine sporadische Mutation in einem einzelnen Spermium zurückzuführen. Eltern, die auf MECP2-Mutationen getestet werden, sind fast immer negativ.
In seltenen Fällen kommt die Mutation jedoch von den Eizellen der Mutter. Folgende beide Szenarien sind möglich.
- Die Eizellen der Mutter könnten die Mutation haben, die als Keimbahnmosaizismus bekannt ist. Ein Nachkomme hätte eine 50prozentige Chance, die Mutation von der Mutter zu erben.
- Die Mutter hat die Mutation in jeder Zelle, aber keine RETT-Symptome wegen der Überlagerung der X-Deaktivierung. Ein Nachkomme hätte wiederum eine 50prozentige Chance, die Mutation von der Mutter zu erben.
Ein Bluttest bei der Mutter würde die überlagernde X-Deaktivierung ausschließen, aber der einzige Weg, den Keimbahnmosaizismus festzustellen, ist eine Analyse der Eizellen.
Eltern eines Sohns mit einer MECP2-Mutation
Der Vater eines betroffenen Sohns benötigt keinen Test, da Jungen das mutierte MECP2-Gen von ihren Müttern erben.
Die Eizellen der Mutter könnten die Mutation haben, die als Keimbahnmosaizismus bekannt ist. Ein Nachkomme hätte eine 50prozentige Chance, die Mutation von seiner Mutter zu erben.
Die Mutter hat die Mutation in jeder Zelle, aber durch eine überlagernde X-Deaktivierung hat sie keine RETT-Symptome. Wiederum hätte ein Nachkomme eine 50prozentige Chance, die Mutation von der Mutter zu erben.
Ein Bluttest bei der Mutter würde die überlagernde X-Deaktivierung ausschließen, doch der einzige Weg, um Keimbahnmosaizismus festzustellen, ist eine Analyse der Eizellen.
Geschwister eines Kindes mit RETT
Das Risiko bei Geschwistern hängt von der genetischen Disposition der Eltern ab.
Wenn man herausfindet, dass die Mutter eines betroffenen Kindes die MECP2-Mutation hat, die auch bei ihrem Kind festgestellt wurde, liegt das Risiko für die Geschwister, die Mutation ebenfalls zu erben, bei 50%.
Wenn eine Mutation bei den Eltern nicht festgestellt wird, ist das Risiko für die Geschwister niedrig. Keimbahnmosaizismus kann dennoch nicht völlig ausgeschlossen werden.
Vorgeburtliche Tests
Vorgeburtliche Tests stehen Eltern mit einem Kind zur Verfügung, bei dem eine MECP2-Mutation festgestellt wurde. Ein vorgeburtlicher Test ist möglich, indem man durch Amniozentese (etwa während der 15. bis zur 18. Schwangerschaftswoche) oder durch Chorionzottenbiopsie (etwa während der 10. bis zur 12. Schwangerschaftswoche) gewonnene DNA analysiert.
Mit freundlicher Genehmigung von http://www.rsrt.org/