http://www.youtube.com/watch?feature=player_embedded&v=_AU5skJVbhY
RETT SYNDROME RESEARCH TRUST WEBSITE
Am 11. November veröffentlichte das renommierte Magazin Cell eine Arbeit von Dr. Alysson Muotri mit dem Titel Ein Modell für Neuralentwicklung und Behandlung des Rett-Syndroms unter Einsatz menschlicher induzierter pluripotenter Stammzellen. Auf dem Gebiet der Stammzellenforschung wurden in den letzten Jahren bemerkenswerte Forstschritte gemacht. Induzierte pluripotente Stammzellen (iPS-Zellen) sind wegen der klinischen Implikationen ein besonders heißes Gebiet. Vereinfacht formuliert erlauben uns iPS-Zellen eine genaue und persönliche Untersuchung von kranken Zellen während ihrer gesamten Lebensdauer. Besonders wichtig ist dabei, dass die in den Zellen aufgefundenen Defizite als ausgelesene Ergebnisse für das Rastern von Medikamenten dienen können.
Ich hatte das Vergnügen, Dr. Muotri vor einigen Jahren kennen zu lernen, um genau zu sein, kenne ich ihn seit seiner Einführung in die Rett-Forschung vor sechs Jahren. Er begann sich für die Störung zu interessieren, während er als Post-Doc im Labor von Fred (Rusty) Gage am Salk Institut in La Jolla, Kalifornien arbeitete. Erfreulicherweise hat sich sein Interesse nicht verändert, und so ist er heute unabhängiger Wissenschaftler an der UCSD.
Dr. Muotri und Kollegen diskutieren ihre Entdeckungen in einem Video (4 min)
Unten finden Sie einen Auszug aus einer Unterhaltung, die Dr. Muotri und ich über diese Arbeit führten.
MC Dr. Muotri, wir gratulieren Ihnen zu Ihrer Arbeit in Cell, die starke Anregungen für die Entwicklung von Medikamenten enthält und daher für alle interessant ist, die ein Kind mit Rett-Syndrom lieben. Ich bin mir bewusst, dass wir in hektischen Zeiten leben. Vielen Dank, dass Sie sich einen Moment nehmen, um mit mir zu sprechen. Ganz neugierig gefragt: Was brachte Sie in die Wissenschaft?
AM Ich wollte schon immer herausfinden, wie die Dinge funktionieren. Entsprechend dachte ich, dass die Wissenschaft der beste Weg ist, um das zu erreichen. Sie wissen, dass ich aus Brasilien stamme. Ich habe meinen Doktor in Genetik an der Universität von São Paulo gemacht. Mein erstes Gebiet war die Krebsbiologie, aber ich schwenkte schnell auf Neurowissenschaften um, als ich 2002 ans Salk-Institut kam. Dort war ich 6 Jahre, bevor ich dann vor zwei Jahren meine aktuelle Stelle hier an der UCSD bekam.
MC Ich nehme an, dass dieser Wechsel von der Krebsbiologie in die Neurowissenschaften eine ziemliche Umstellung war.
AM Ja, am Anfang hat mich das ein bisschen eingeschüchtert, weil es so viel zu lernen gab. Aber ich nahm die Herausforderung an und es stellte sich heraus, dass meine Erfahrungen in der Krebsforschung sehr vorteilhaft für den Wechsel zu den Neurowissenschaften waren. Als ich beispielsweise in Rustys Labor kam, war eine unserer ersten Beobachtungen mit einem Phänomen verbunden, das Transposons heißt. Ich wusste aus meiner vorherigen Arbeit, dass Retrotransposons in Krebszellen sehr aktiv sind und habe das mit meinen Kollegen aus den Neurowissenschaften diskutiert. Die meisten von ihnen waren mit diesem Phänomen nicht vertraut und hielten es für unbedeutend. Ich dagegen hatte das Gefühl, dass es besser wäre, sich diese Transpositionen genau anzusehen, wenn sie denn wirklich im Gehirn stattfinden. Schließlich könnte das an einem neuen Mechanismus, der mit der Hirnentwicklung zusammenhängt, beteiligt sein.
MC Da Retrotransposons das Thema Ihrer nächsten Arbeit über Rett ist, die demnächst in Nature erscheint, geben Sie mir kurz Gelegenheit, unsere Leser mit ein paar Hintergrundinformationen zu versorgen. Retrotransposons sind DNA-Sequenzen, die sich bewegen und an neuen Stellen im Genom einbauen. Barbara McClintock erhielt 1983 den Nobelpreis für die Entdeckung dieses Phänomens. In der Vergangenheit wurden Retrotransposons als „Ramsch-DNA“ betrachtet, weil sie rund 50 % des Genoms von Säugetieren besetzen und keine eindeutige Funktion innerhalb der Zelle haben. Vielleicht ist es wahrscheinlicher, dass Retrotransposons eine biologische Funktion haben, die uns im Moment noch unklar ist. Retrotronsposons wurden schon mit Krankheit in Verbindung gebracht.
Dr. Muotri, würde Sie uns vielleicht einen Einblick in Ihre bald erscheinende Arbeit gestatten, wo Sie sich mit Retrotransposons beim Rett-Syndrom beschäftigen?
AM Die Idee ist, dass Retrotransposons, die herumspringen und sich in das Genom einbauen, innerhalb des gleichen Individuums an der Ausbildung von Nervenzellen beteiligt sind, die sich genetisch voneinander unterscheiden. Außerdem haben wir beobachtet, dass MECP2, das am Rett-Syndrom beteiligte Gen, diese Aktivität vorrangig unterdrückt. Dann haben wir festgestellt, dass diese Sprünge fast ausschließlich im Gehirn stattfinden und dass MECP2 einer der Torwächter zu sein scheint, der die Stärke dieser Aktivität kontrolliert.
MC Das bedeutet, dass in einem Gehirn mit einem defizitären MECP2-Protein verstärkt solche Sprünge stattfinden?
AM Genau. Wir müssen allerdings noch sehen, ob diese Extraaktivitäten zu den Rett-Symptomen beitragen, oder ob das Gehirn einfach kompensiert und quasi um sie herum arbeitet. An dieser Frage arbeiten wir gerade.
MC Das klingt spannend. Ich freue mich sehr, unser Gespräch fortzusetzen, wenn sie weitere Fortschritte gemacht haben. Zwei Spitzenarbeiten in einem Monat – das ist schon sehr beeindruckend.
Doch zurück zu iPS. Auf diesem Gebiet wurden innerhalb kurzer Zeit bemerkenswerte Fortschritte erzielt. Können Sie unseren Lesern das Spannende an diesen Zellen erklären?
AM Der Traum der Neurowissenschaftler ist ein Verständnis der frühen Stufen einer neurologischen Störung. Bisher hatten wir zwei Möglichkeiten, dies zu erreichen. Bei der einen entwickelt man ein Mausmodell, das hoffentlich die beim Menschen vorhandenen Symptome spiegelt. Natürlich liegt die Begrenzung eines solchen Modells darin, dass es sich eben um eine Maus handelt und nicht um einen Menschen – dessen Gehirn ja um einiges komplexer ist. Die andere Möglichkeit ist postmortales Hirngewebe. Hier ist das Problem, dass die Schädigung zu dieser Zeit schon stattgefunden hat und dass man die letzte Stufe der Krankheit sieht. Um ein Leiden wirklich zu untersuchen ist es sehr hilfreich, die denkbar primitivste Reihe von Zellen zu haben und diese Zellen dann in eine Vielfalt verschiedener Zellkulturen zu übertragen. Auf diese Weise kann man untersuchen, was zu verschiedenen Zeitpunkten passiert. Ein wichtiger Durchbruch, der diese Arbeit sehr machbar erscheinen ließ, wurde vor einigen Jahren erreicht. Ein japanisches Forscherteam unter der Leitung von Sinya Yamanaka überraschte die Welt, als sie zeigte, dass man Zellen, die bereits zu einem naiven Status rückdifferenziert haben, der an den der menschlichen embryonalen Stammzelle erinnert, neu programmieren kann. Dies erlaubt uns, das Genom einer Person einschließlich jedweder genetischen Mutation zu erfassen und die Nervenzellen oder andere, für die wir uns interessieren, zu untersuchen. So können wir sehen, wie eine Krankheit voranschreitet und welche Veränderungen auf der molekularen Ebene stattfinden.
MC Bisher hat sich die Rett-Forschung vornehmlich auf die Mausmodelle als Herangehensweise verlassen. Das ist im Bezug auf die Medikamentenrasterung eine sehr teure Herangehensweise. Die Verfügbarkeit von iPS-Reihen mit MECP2-Mutationen erlaubt den Forschern einen zellulären Ansatz, um nach Wirkstoffen zu rastern. Tausende oder gar Hunderttausende von Inhaltsstoffen können so schnell und effizient in Zellreihen gerastert werden, wobei sowohl Gering- oder Hochdurchsatztechnologien zum Einsatz kommen können.
Können Sie uns etwas über die Phänotypen sagen, die Sie in den Zellen gefunden haben? (Ein Phänotyp ist ein sichtbares Charakteristikum oder eine Eigenschaft)
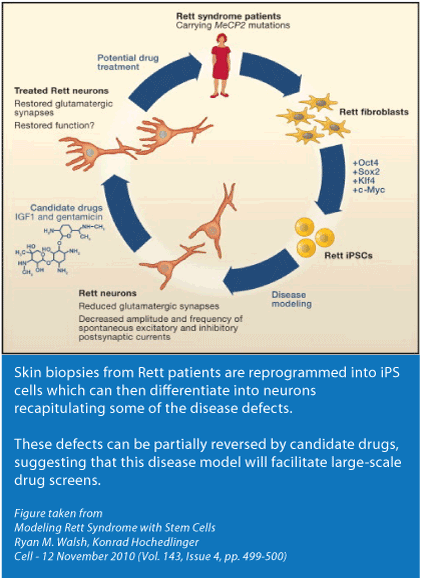
Hautbiopsien von Rett-Patienten werden zu iPS-Zellen rückprogrammiert, welche dann wiederum zu Nervenzellen differenzieren können. Dabei behalten diese einige der krankhaften Schäden. Diese Schäden können teilweise mit denkbaren Medikamenten behoben werden, in der Annahme, dass diese Krankheitsmodelle groß angelegte Medikamentenraster ermöglichen.
Abbildung aus: Modelling Rett Syndrome with Stem Cells
Ryan M. Walsh, Konrad Hochedlinger
Cell – 12. November 2010 (Band 143, Ausgabe 4, S. 499-500)
AM Einer der Phänotypen hing mit der Zellsomagröße (Zellkörper) einer Nervenzelle zusammen. Einfach indem wir Nervenzellen unter dem Mikroskop ansahen, fanden wir heraus, dass Rett-Nervenzellen ungefähr 10 % kleiner sind. Das sieht nach nichts Besonderem aus, aber wenn man die dreidimensionale Struktur einer Nervenzelle berücksichtigt, ist eine 10%ige Verringerung schon bedeutsam. Die Größe war also das einfachste ausgelesene Ergebnis, das wir gefunden haben.
Ein anderer Phänotyp steht im Zusammenhang mit der Morphologie der Nervenzelle. (Morphologie ist die Untersuchung der Struktur und Form eines Organismus.) Die Idee, genauer auf die Morphologie zu schauen, entstand durch die Berichte über postmortale Hirngewebe von Menschen und Tiermodellen aus dem letzten Jahrzehnt. Wir konzentrierten uns auf die Spinaldichte bei Nervenzellen und sahen dort ebenfalls eine Verringerung. (Ein Dendritenspinal, oder einfach Spinal, ist eine kleine membranartige Ausbuchtung des Dendriten einer Nervenzelle, wo typischerweise der Input einer Synapse stattfindet.) Wir haben uns neuronale Netzwerke angesehen und Defizite in deren Kommunikationsfähigkeit gefunden.
MC Sie haben iPS-Zellen mit unterschiedlichen MECP2-Mutationen entwickelt. Dabei haben Sie herausgefunden, dass die Phänotypen bei den Mutationen durchgängig vorkamen. Können Sie das weiter ausführen?
AM Ja, die vier verschiedenen Mutationen, die wir untersucht haben, führten zu ähnlichen Phänotypen. Zumindest bei den Phänotypen, die wir uns angesehen haben. Und so waren wir überzeugt, dass dies einen Rückschluss auf einen Verlust der MeCP2-Funktion mehr als nahe legt. Also schalteten wir die MeCP2-Expression bei Kontrollnervenzellen aus und kamen zum gleichen Ergebnis. Dann stellten wir das normale MeCP2-Gen in Rett-Nervenzellen wieder her, so dass die Phänotypen unterdrückt wurden. In Kombination legen diese Experimente die Annahme nahe, dass MeCP2 für die Veränderungen bei Rett-Nervenzellen verantwortlich ist. Die Tatsache, dass verschiedene MeCP2-Mutanten einen ähnlichen Phänotyp hervorbrachten, ist klinisch relevant, weil sie darauf hindeutet, dass möglicherweise ein einzelnes Medikament alle gemeinsam korrigieren kann.
MC Das Ziel wäre demnach, diese Zellen aus Plattform für Medikamentenraster zu benutzen.
AM Richtig. Als Prinzipienbeweis fügten wir den Zellen einen Wachstumsfaktor, IGF 1, hinzu. Wie Sie wissen, wurde Anfang 2009 eine Arbeit in PNAS veröffentlicht, die zeigte, dass ein Wirkstoff, der IGF 1 ähnlich ist, einige der Symptome bei Mausmodellen verbesserte. Entsprechend beschlossen wir, das in unserem System auch zu probieren. Dabei kam heraus, dass IGF 1 den Phänotyp korrigierte, ja sogar überkorrigierte. Diese Überkorrektur muss im Zusammenhang mit klinischen Versuchen beachtet werden, wünschenswert wäre natürlich eine ordentliche Abstimmung der Dosierung bei jedem Patienten. Außerdem sollte man im Kopf behalten, dass das IGF 1 bei klinischen Versuchen ins Gehirn gelangen muss, und dass passiert nicht so, wie wir es gern hätten. Jetzt bringen wir das IGF 1 ja direkt in die Zellen.
Das andere Medikament, das wir ausprobiert haben, ist Gentamicin, ein Antibiotikum, das vorreife Stoppkodons „durchlesen“ kann (Nonsensmutationen, die mit X enden, wie etwa 255X, 168X). Wir fanden heraus, dass Gentamicin Stufen des MeCP2 wieder herstellte und die Zellen phänotypisch rettete.
MC Das ist ziemlich interessant, besonders wenn man bedenkt, dass durchlesende Medikamente wirken, indem sie das Stoppkodon durch eine beliebige Aminosäure ersetzen. Also ersetzt man im Endeffekt eine Mutation durch eine andere.
AM Wir haben das untersucht und gesehen, dass die Proteinstufe normal war. Wir hatten aber keine Möglichkeit zu sehen, welche Mutation eingebaut wurde. Ein Teil des neuen Proteins, das in Gegenwart von Gentamicin zusammengesetzt wird, ist vielleicht in Ordnung, und wir glauben, dass genau dies die Phänotypen umkehrt.
MC Man sollte nicht vernachlässigen, dass Gentamicin hochgiftig ist und die Blutschranke im Hirn nicht gut passiert. Es kann also momentan keineswegs zur Behandlung des Rett-Syndroms dienen. Doch es gibt andere Medikamente mit ähnlichen Wirkweisen, die momentan an Tiermodellen getestet werden.
Aber die Botschaft, die wir aus Ihren Daten mitnehmen können ist, dass iPS-Zellreihen ein in vitro-Modell für das Rett-Syndrom darstellen und entsprechend als Plattform für Medikamentenraster verwendet werden können. Was sind die nächsten Schritte, um iPS-Reihen auf diese Weise einzusetzen?
AM Der nächste Schritt ist, diesen Rahmen zu vergrößern, und das ist nicht leicht. Das vor allem, weil die Experimente sehr anfällig für Variable sind. Außerdem gibt es viele Schritte bei der Umwandlung von iPS-Zellen zu Nervenzellen. Wir müssen also alle Variablen systematisch validieren und die Systeme so zuverlässig wie möglich machen. Schließlich müssen wir die angemessenen ausgelesenen Ergebnisse auswählen (die zellulären Phänotypen), die wir verwenden wollen. Es ist dabei sehr wichtig, diese Experimente behutsam anzulegen, damit man keine Zeit mit Falschmeldungen verschwendet. Mein Labor hat kürzlich eine CIRM-Finanzierung erhalten (California Institute for Regenerative Medicine; Kalifornisches Institut für regenerative Medizin), genau um diese Schritte zu optimieren. Deshalb würde ich gern so bald wie möglich anfangen. Letztendlich würde ich gern ganze Batterien von Medikamenten testen, die vorher durch klinische Tests für andere Krankheiten gefallen sind. Repositionierung von Medikamenten, so nennt man dieses Konzept, ist sehr attraktiv, weil Medikamente mit einer neuen Einsatzabsicht viele der frühen Kosten nicht verursachen und auch nicht soviel Zeit gebraucht wird, um das Medikament auf den Markt zu bringen.
MC Für mich ist Ihre Arbeit aus zahlreichen Gründen sehr ermutigend. Zunächst bestätigen und bestärken Ihre Daten weiterhin die Idee, dass Rett reversibel ist. Zweitens haben Sie gezeigt, dass die iPS-Plattform sich für Medikamentenraster eignet. Drittens lassen Ihre Daten vermuten, dass die möglicherweise zahlreichen Mutationen auf MECP2 gemeinsame Phänotypen teilen. Dies kann ein bedeutender Ansatz im Zusammenhang mit Behandlungsstrategien sein.
Dr. Muotri, ich denke, ich spreche für jede Rett-Familie, die dieses Interview liest. Wir wünschen Ihnen viel Glück und Erfolg bei Ihrer Arbeit.