Meilensteine
Meilensteine
Im Jahr 2017 wurde ein dreijähriger strategischer Forschungsplan, die „Roadmap to a Cure“, umgesetzt. Ziel war es, therapeutische Therapieprogramme zu identifizieren und einzuleiten, die auf die Grundursache des Rett-Syndroms abzielen.
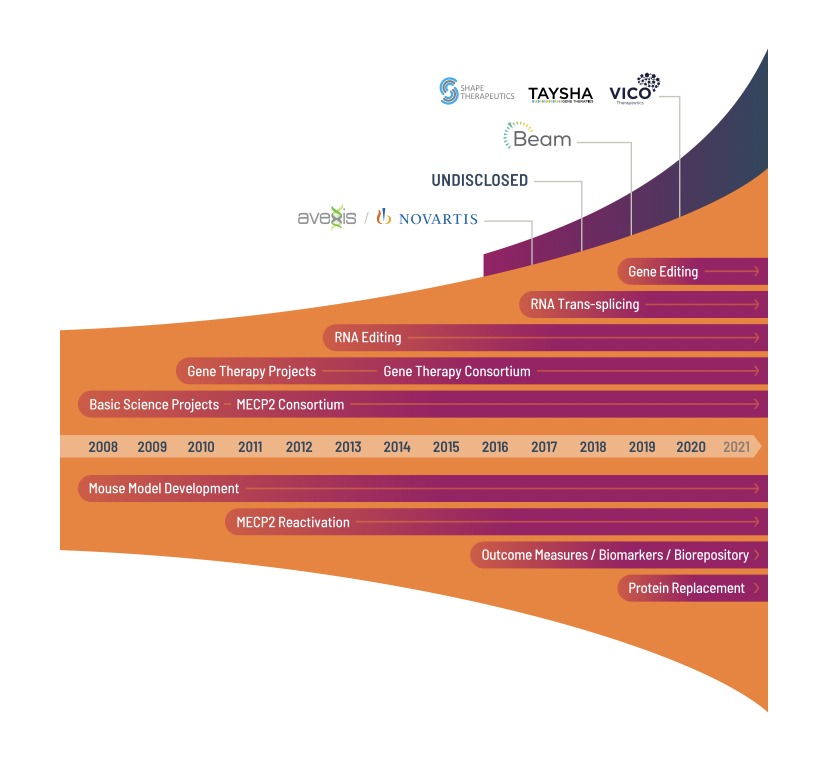
Als die Roadmap begann, befanden sich drei MECP2-Therapieprogramme in der Pipeline. Die Roadmap erleichterte in Verbindung mit früherer Unterstützung den Start von 12 weiteren Programmen. Bemerkenswert ist, dass es zu Beginn der Roadmap NULL Unternehmen mit MECP2-Therapieprogrammen gab. Heute gibt es SECHS, und weitere werden folgen.

THERAPEUTISCHE ENTWICKLUNG
- Der RSRT führte neue therapeutische Bereiche für Rett ein und erweitert die, die vorher nicht existierten: Gentherapie, Genom-Editing, MECP2-Reaktivierung, RNA-Editing, RNA-Trans-splicing.
- Die von RSRT angestoßene Forschung hat dazu geführt, dass die Pharmaindustrie Programme aufgreift: AveXis, Taysha, Vico.
- Von RSRT unterstützte Forschungs- und Grundlagen Fakten haben Programme beschleunigt: Beam, Shape.
- Entwicklung eines Gentherapie-Austauschprodukts der nächsten Generation unter Beteiligung des Gentherapie-Pioniers Jim Wilson.
- Aufbau, Rekrutierung und Finanzierung eines Weltklasse-Teams an der UMASS Medical School zur Bearbeitung der DNA durch den Ersatz von Abschnitten, die 97% aller Mutationen enthalten.
- Fünf der weltbekannten Experten, darunter einen Erfinder von CRISPR, für die Arbeit an der RNA-Editierung sind rekrutiert worden.
- Durch unsere Finanzierung wurden drei Ansätze zur Regulierung der MECP2-Expression entwickelt, um eine Überexpression in der Gentherapie zu verhindern.
- Das MECP2-Konsortium, das sich aus einem Elite-Wissenschaftler-Trio zusammensetzt, hat die wissenschaftliche Denkweise darüber, wie MECP2 funktioniert, umgestaltet.
RETT SYNROME RESEARCH TRUST ROLLE ALS GRÜNDUNGSZENTRUM
Die grundlegende Wissenschaft, die Übertragungsbemühungen, Werkzeuge und Ressourcen, die wir gefördert und unterstützt haben, haben es den unten aufgeführten Biopharmaunternehmen erleichtert, MECP2-Therapieprogramme zu initiieren. Zu Beginn der Roadmap to a Cure gab es NULL-Firmen. Jetzt gibt es SECHS, und weitere werden folgen.
KLINISCHE UND TRANSLATIONALE FORSCHUNG
- Startete die Outcome Measures & Biomarkers Initiative, um Auswertungen zu entwickeln, die aussagekräftig und für die FDA akzeptabel sind.
- Konzeption und Gründung eines Konsortiums für klinische Studien mit dem Ziel, Ressourcen für die Durchführung qualitativ hochwertiger klinischer Studien bereitzustellen.
- Im Jahr 2019 startete unsere erste von der FDA regulierte Studie im Rahmen des Konsortiums für klinische Studien.
- Schaffung eines Biorepositoriums mit wertvollen menschlichen Rett-Gewebeproben (Fibroblasten, Lymphozyten, iPSC), die der wissenschaftlichen und biopharmazeutischen Forschung zur Verfügung stehen.
- Startete die Maus-Modell-Initiative der Jackson Laboratories.

History of Roadmap to a Cure
Der RSRT hat drei Ansätze zur Heilung des Rett-Syndroms identifiziert und Projekte dazu in Auftrag gegeben.
Das menschliche Genom enthält 30,000 individuelle Gene. Ein Gen kann welliges Haar oder blaue Augen kodieren, aber auch die Anfälligkeit oder Abwehrkraft für/gegen Krankheiten. Es gibt darüber hinaus eine andere Art von Genen, die im zurzeit an Interesse gewinnenden Feld der Epigenetik untersucht werden. Hier handelt es sich um Mastergene, die eine Kontrollfunktion haben und andere Gene im Zusammenspiel mit Entwicklungsgegebenheiten oder Umweltfaktoren ein- und ausschalten, um normales Wachstum und Gesundheit zu gewährleisten. Epigenetik beeinflusst die Forschung auf sämtlichen Gebieten, von Krebs bis hin zu psychischen Störungen.
Mutationen an einem Gen mit dem Namen MECP2 sind der Auslöser des Rett-Syndroms. MECP2 kontrolliert viele andere Gene. Aktuelle Arbeiten nehmen an, dass es direkt oder indirekt die Aktivität von möglicherweise Tausenden anderen Gene regelt, die ordentlich koordiniert sein müssen, damit ein korrekt funktionierendes Gehirn und Nervensystem vorhanden sind. Die Wiederherstellung der entsprechenden Stufen des MECP2 hat nachweislich dazu geführt, dass Schäden, die durch eine mutierte Kopie des Gens aufgetreten sind, rückgängig gemacht werden konnten. Dies zeigt uns den enormen Einflussbereich von MECP2, weil ein Symptom nach dem anderen in vollständig ausgewachsenen Modellen für das Rett-Syndrom verschwunden ist.
Dieser erstaunliche vorklinische Durchbruch führt uns zu der dringenden Herausforderung herauszufinden, ob solche Ergebnisse bei Menschen, die am Rett-Syndrom leiden, ebenfalls erreicht werden können. Die Neurobiologie im Zusammenhang mit dem Rett-Syndrom erweist sich als komplex, und die Funktion des MeCP2-Protein bleibt schwer zu fassen. Grundlagenforscher in aller Welt konzentrieren sich weiter auf solche Forschungsansätze.
Während diese wichtige Arbeit vorangeht, gibt es deutliche Ansätze im Hinblick auf Intervention, die parallel verfolgt werden sollten, um die Funktion von MECP2 zu verstehen. Mit ihrer Erfolgsbilanz für raschen Fortschritt haben die Berater und Leiter des RSRT drei solche Ansätze zur Behandlung und Heilung des Rett-Syndroms identifiziert.
Steigerung der Stufen des MeCP2-Proteins
1. Das Rett-Syndrom wird durch eine Schwäche des MeCP2-Proteins ausgelöst. Ein Ansatz zur Heilung der Störung ist daher, normale Stufen wiederherzustellen. Dies kann auf zahlreichen Wegen erreicht werden, einschließlich kleinerer Molekültherapeutika (Medikamente) und/oder biologischer Wirkstoffe (Gentherapie, Austausch von Proteinen).
Einige Behandlungsweisen könnten sich als mutationsspezifisch herausstellen (etwa Medikamente, die auf die Wiederherstellung der normalen Funktion eines abnorm gekürzten Proteins abzielen, oder Medikamente, die fehlgefaltete Proteine neu zusammensetzen), während andere allgemeiner wirken mögen, etwa indem sie das normale MECP2-Gen auf dem stummen X-Chromosom aktivieren.
Der Trust finanziert gegenwärtig eine umfassende Palette an Projekten auf diesem Gebiet. Das erste, durchgeführt im Labor von Dr. Antonio Bedalov, zielt darauf ab, das MECP2-Gen auf dem stummen X-Chromosom zu aktivieren. Ein zweites Projekt findet im Labor von Dr. Marisa Bartolomei statt. Sie sucht nach den grundlegenden Mechanismen, die das stumme MECP2-Gen auf dem inaktiven X-Chromosom abgeschaltet halten. Ein weiteres Projekt am Labor von Dr. Stavros Lomvardas untersucht Medikamente mittels Hochdurchsatz-Rasterung, um möglicherweise einige zu finden, die das Defizit umkehren können, das kürzlich an olfaktorischen Rezeptornervenzellen (ORN) bei einer MeCP2-Schwäche entdeckt wurde. Zwei Gentherapieprojekte werden am Labor von Ronald Crystal und Brian Kaspar durchgeführt. Sie verwenden verschiedene Vektoren (Trojanische Pferde), um MECP2-Gene zu in den Organismus einzubringen. Wir hoffen in ein paar Jahren zu wissen, ob Gentherapie eine erfolgversprechende Wahl für das Rett-Syndrom ist. Adrian Bird, ein Kollege von Crystal, arbeitet ebenfalls an zwei Ansätzen zur Stärkung von MeCP2.
Linderung spezifischer Symptome
2. Das Spektrum einzelner Symptome beim Rett-Syndrom ist so erheblich, dass bereits das Ausschalten eines einzigen in vielen Fällen die Lebensqualität entscheidend verbessern kann. Ein vom FDA (Arzneimittelbehörde in den USA, d.Ü.) zugelassenes Medikament/einen Wirkstoff zu finden, das/der ein Symptom behandeln kann (etwa Atemstörungen, extreme nervöse Angst, Krämpfe), wäre der schnellste und kostengünstigste Weg zu klinischen Versuchen.
Dr. Andrew Pieper wird mit Unterstützung des RSRT existierende Wirkstoffe an einem Tiermodell der Krankheit rastern. Der RSRT unterstützt ein Projekt am Labor von Huda Zoghbi, wo eine detaillierte Tiefenevaluation von spezifischen, vom FDA zugelassenen Medikamenten durchgeführt wird. Jonathan Kipnis experimentiert daran, das Immunsystem bei Rett über Knochenmarkstransplantationen und pharmakologische Interventionen zu stärken.
Identifizierung von Zielgenen und Genen, die MECP2-Mutationen verändern
Die Entwicklung von Interventionsmöglichkeiten bei Genen, die von MECP2 kontrolliert werden, stellt einen weiteren potenziellen Weg dar. Aktuelle Daten lassen nämlich vermuten, dass MECP2 möglicherweise Tausende Gene kontrolliert. Außerdem könnten diese Gene auf bemerkenswerte Weise variieren, abhängig vom jeweiligen Gewebetyp. Es wäre daher eine schwierige Herausforderung, Behandlungsmöglichkeiten zu entwickeln, wenn Tausende von Genen angepeilt werden müssen. Trotzdem ist die Identifizierung dieser Gene das zentrale Interesse einiger Labore, und der Fortschritt auf diesem Gebiet mag zu Genen führen, die man sinnvollerweise weiter verfolgen kann.
Eine vielleicht noch interessantere Herangehensweise ist die Untersuchung von MECP2-Modifikatorengenen. Es ist nicht unwahrscheinlich, dass Unterschiede in der genetischen Disposition eines einzelnen Menschen die Auswirkungen einer MECP2-Mutation verändern können. Es gibt Menschen mit gewöhnlichen MECP2-Mutationen und einer normalen Deaktivierung des X-Chromosoms, die aber kein Rett-Syndrom haben. Wahrscheinlich werden diese Menschen durch Mutation(en) an anderen Genen vor ihrer MECP2-Mutation geschützt. Die Identifizierung dieser Modifikatorengene könnte neue Wege zur Behandlung eröffnen.
Ein Projekt am Labor von Dr. Monica Justice versucht MECP2-Genmodifikatoren zu identifizieren.
Mit freundlicher Genehmigung von http://www.rsrt.org